Spinal Muscular Atrophy (SMA)
About SMA
Spinal Muscular Atrophy (SMA) refers to a group of diseases which affect the motor neurons of the spinal cord and the brain stem. These are very important cells that are responsible for sending signals to the muscle cells. Without proper input from the motor neuron, the muscle cells will become much smaller (atrophy) and will produce symptoms of muscle weakness. Degeneration and death of the motor neurons in the key organs such as brain and spinal cord leads to weakness in the muscles connected with swallowing, breathing and walking. The incidence of SMA is between 1 in 6000 to 1 in 10,000 live births with a carrier frequency of 1 in 50 all over the world. There are 4 types of SMA which can be categorized according to the age of onset, mode of inheritance, distribution of muscle weakness and progression of symptoms. The most common cause of SMA is due to deficiency of a motor neuron protein called SMN (Survival Motor Neuron).
Inheritance of SMA
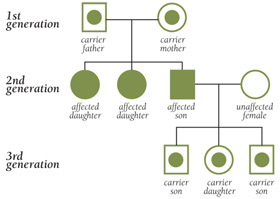
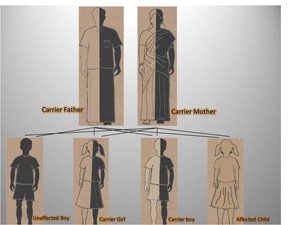
For a child to develop SMA, both the parents should be possessing the defective gene which will be later on passed to their child. These parents are known as carriers, and don’t show symptoms of SMA. In a case where both the parents are SMA carriers and give birth to a child, there is a 50% chance that the offspring will be a carrier of SMA and 25% chance that the offspring will develop SMA. This is called autosomal recessive inheritance.
Symptoms of SMA
The symptoms of SMA vary with stages. Some of the common symptoms that manifest in the different types are mentioned below
Type I (Infantile): Werdnig – Hoffmann Disease (Age of onset: 0 – 6 Months)
1. Hypotonia
2. Diminished muscle tone
3. Muscle weakness
4. Respiratory weakness
5. Swallowing and feeding difficulties
6. Weak cry
7. Quivering of the tongue
8. Unable to sit without support
9. Life expectancy- They usually may not live beyond 2 years
Type II (Intermediate SMA): Dubowitz disease (Age of onset: 6 – 18 months)
1. Decreased or absent deep tendon reflexes, the reflex that occurs when you tap on your knee
2. Hypotonia or diminished muscle tone
3. Contractions or twitching of muscles called fasciculations
4. Able to sit without support, but unable to walk without support
Type III (Kugelberg – Welander Disease or Mild / Juvenile SMA) (Age of onset: after 18 months of age)
1. Rarely experience respiratory or swallowing problems
2. Weakness in shoulders, hips, thighs
3. Able to walk without support for sometime although many later lose this ability
Type IV (Adult – onset SMA)
1. Usually manifests after the third decade of life with gradual weakening of muscles
2. Mainly affects proximal muscles of the extremities – frequently rendering the patient wheelchair-bound
3. Other complications are rare, and life expectancy is unaffected
Diagnosis of SMA
Diagnostic evaluation for Spinal Muscular Atrophy is shown in the figure below. The Standard of Care Committee (to treat NMD) recommends the following step-wise diagnostic procedure when encountering the patients with clinical symptoms of SMA. Briefly, the first diagnostic test for a patient suspected to have SMA should be the SMN gene deletion test. A homozygous deletion of the SMN1 gene exon 7 (with or without deletion of exon 8) confirms the diagnosis of SMN-associated SMA (5q-SMA). The other diagnostic tests should be ordered only after a negative SMN gene test has been obtained.
Management of SMA and the future of SMA research
Management of SMA is focused on life support, encouraging independence and/or improving the patient’s quality life. Care for the patients with SMA should be tailored according to their current functional status rather than the original classification of SMA types. A multidisciplinary approach towards care and treatment is required for optimal comfort of children. Examples of care and treatment include:
1. Family education and counseling on the disease process, pathogenesis, prognosis etc.
2. Multidisciplinary intervention including pediatric neuromuscular clinics, genetics and diet
Physiotherapy
3. Use of assistive devices such as wheelchairs, breathing machines and feeding tubes.
4. Surgery for spinal deformity
5. Management of respiratory disorders using medication and equipment
6. Information for reproductive planning (prenatal or pre-implantational diagnosis)
Research is progressing rapidly to find a cure for SMA. Since cure is not available yet, prevention and management is the only way to handle the disease. Respiratory drainage and physiotherapy is necessary for the patients to improve their comfort and standard of living. Antenatal screen test with the use of chorionic villus sampling or CVS is also available for identifying the gene. Lack of SMN1 homologue of the gene is identified as the cause of this disease. For this reason, researchers are searching for drugs that could increase the copy number of SMN2 and maintain the functions of SMN gene. SMA is a model for translational research because it is closest to a treatment or cure out of all 300 neurological disorders.
Ongoing research for SMA
The FightSMA research strategy attacks SMA on six tracks
1. Trick the remaining SMN2 gene into producing more protein.
2. Replace the missing gene.
3. Replace the protein.
4. Find a small molecule substitute.
5. Administer protective or growth-enhancing agents.
6. Grow new motor neurons from stem cells.
Clinical trials
High throughput screens by various research teams have resulted in leads on possible compounds that may
1. Upregulate production of SMN protein by the SMN2 gene
2. Improve transcription of SMN protein by the SMN2 gene, thus improving “yield”.
Compounds identified by the various screens include some that are already FDA approved. Clinical trial of one or more of these compounds may be initiated soon.
We Offer Following Services For Spinal Muscular Atrophy
1. Diagnostics
2. Counseling
3. Registry
4. Outreach